Introduction
Dermatomyositis, a rare autoimmune disorder affecting muscles and skin, typically manifests with a gradual onset of symptoms. Prominent skin features include a reddish-purple peri-orbital rash, knuckle papules, and a distinctive V-shaped upper back and chest rash, with symptom variation among individuals. Dermatomyositis can affect multiple organ systems, with cardiac involvement significantly contributing to morbidity and mortality. Historical awareness of cardiovascular implications dates back to Oppenheim's observations in 1899, with formal documentation by Bohan et al. in 1977, reporting cardiovascular abnormalities in 19% of polymyositis and 6% of dermatomyositis patients [1].
Literature underscores arrhythmias and congestive heart failure (prevalent in 10-15%) as common cardiac complications in dermatomyositis [2]. Dermatomyositis patients face an increased risk of atherosclerosis development, with case reports documenting severe systolic heart failure and long-term diastolic dysfunction in adults with idiopathic inflammatory myopathies [3]. Proposed mechanisms include myocarditis, characterized by myocardial inflammation similar to that in skeletal muscles, whereas pericarditis is often asymptomatic and clinically insignificant. Early diagnosis necessitates a cardiac symptom and history evaluation, with essential diagnostic tools including electrocardiography (12-lead ECG, 24-hour ambulatory ECG, 2D echocardiograms, and M-mode echocardiograms) for detecting ST-T changes, arrhythmias, and conduction abnormalities. Frequent ECG changes require prompt treatment. Technetium 99m-pyrophosphate scintigraphy can identify left ventricular abnormalities, reduced function, hyperkinetic contraction, and systolic dysfunction. Cardiac troponin I (cTnI) serves as a specific marker for myocardial injury.
Treatment strategies, evolving from case reports and series, encompass Disease-modifying antirheumatic drugs (DMARDs) combined with immunosuppressants for idiopathic inflammatory myopathies with myocarditis. Standard cardiac medications, including beta-blockers, calcium channel blockers, angiotensin-converting enzyme inhibitors, nitrates, and diuretics, address heart failure, cardiomyopathies, and arrhythmias. Severe atrioventricular blockages may require pacemaker implantation. Hypertensive and dyslipidemic patients receive HMG Co-A reductase inhibitors (statins), beta-blockers, and aspirin. Caution is necessary when administering statins in idiopathic inflammatory myopathies due to potential myopathy exacerbation.
Case Report
We hereby report a 26-year-old male patient with no known co-morbidities who initially complained of a blackish-red rash on his forearm, malar area, and difficulty rising from a seated position [Fig.1]. A muscle biopsy revealed mild inflammation in the perimysium and endomysium, with a myositis profile confirming inflammatory dermatomyositis (Mi 2+ subtype). Treatment commenced with oral steroids and azathioprine, resulting in noticeable improvement over a three-month period. However, the patient discontinued the medications.
In 2020, the patient experienced a recurrence of myositis symptoms and skin rashes. Additionally, he developed hard swelling over his elbows, chest, lower abdomen, and bilateral anterior thighs, accompanied by extensive and deep calcinosis cutis [Fig.1]. From April 2021 onwards, the patient's symptoms worsened. Investigations revealed elevated creatine phosphokinase (CPK) levels, low-normal creatinine, and anemia indicative of chronic illness. Initially, he was administered intravenous cefoperazone plus sulbactam for fever and received treatment for severe hypoalbuminemia and pedal edema with human albumin and diuretics. A 2D echocardiogram revealed cardiomyopathy with moderate left ventricular (LV) dysfunction (EF: 43%), for which he was prescribed torsemide, spironolactone, ramipril, and bisoprolol.
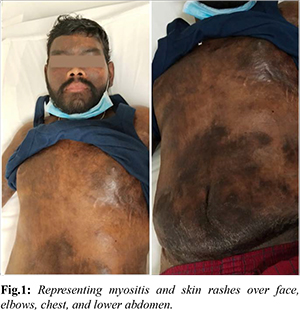
Due to the presence of inflammatory polyarthritis, muscle weakness, and extensive calcinosis, the patient received low-dose oral steroids, non-steroidal anti-inflammatory drugs (NSAIDs), weekly methotrexate, and Rituximab. Following hospitalization, the patient's condition stabilized. Two months later, during a cardiology outpatient visit, his condition had significantly improved. He reported no angina or shortness of breath and was able to resume regular activities. ECG and 2D echocardiography revealed global hypokinesia with an improvement in LV dysfunction from moderate to mild (EF: 50%) and a global longitudinal strain (GLS) of LV 16%, which subsequently improved to 18%. He continued the same medication regimen for another two months, during which his condition further improved. Follow-up myocardial scans (perfusion scans) demonstrated good perfusion in all segments.
Discussion
Cardiac involvement in polymyositis and dermatomyositis is a frequent and concerning issue, often associated with poor prognostic outcomes. The insidious and delayed onset of cardiac symptoms can lead to under-recognition by both patients and physicians. A comprehensive review by Zhang Lu et al. highlighted that heart failure is the most commonly reported complication in dermatomyositis patients. Among dermatomyositis patients, clinical evidence of cardiac involvement, characterized by heart failure with symptoms like dyspnea on exertion, orthopnea, and paroxysmal nocturnal dyspnea, is prevalent in 32% to 72% of cases out of 1530 patients. Conduction abnormalities were observed in 25-38% of patients, with the incidence of left ventricular diastolic dysfunction and hyperkinetic left ventricular contraction at 42% and 6-12%, respectively. Alarmingly, 46.3% of these patients succumbed to mortality due to heart disease [4]. Another review article by Divya Jayakumar delved into the prevalence of cardiac complications in polymyositis and dermatomyositis, revealing a wide range of prevalence rates (6-75%) dependent on the type of cardiovascular involvement (subclinical or symptomatic). Interestingly, 13-72% of cases remained asymptomatic. Mortality rates varied from 5% to 48%, contingent on factors such as gender (male), older age, cardiopulmonary involvement, and cancer, as well as follow-up time [5].
Current treatment guidelines draw from documented case reports and series. Allanore et al. reported successful treatment approaches, including steroid therapy followed by a combination of steroids and disease-modifying antirheumatic drugs (DMARDs). Intravenous pulse steroids, prednisone, IV cyclophosphamide, hydroxychloroquine, azathioprine, and prednisone have all been employed based on the specific patient and condition. Yoshimatsu et al. also highlighted the use of intravenous immunoglobulins (IVIG) in resolving Dermatomyositis-associated cardiomyopathy [6,7].
Despite cardiac involvement in myositis often remaining clinically subtle, it remains a significant contributor to disease morbidity and mortality. As a result, we recommend that all patients diagnosed with Idiopathic Inflammatory Myopathies, including those in remission, undergo regular cardiac function assessments to mitigate cardiovascular risks. A comprehensive strategy for monitoring and intervention is essential.
Conclusion
Early detection of cardiac muscle inflammation through precise diagnostic tests can enable timely initiation of therapy, potentially reducing mortality rates associated with these conditions. It is crucial to acknowledge that both the incidence and prevalence of myositis-related cardiac complications are likely underreported, necessitating more robust data collection and awareness-building efforts. In the pursuit of improving patient outcomes, clinical trials in this field are imperative to develop novel and enhanced treatment modalities aimed at mitigating the risks associated with these diseases.
Contributors: KF, SM: Collection and compilation of data, and writing of initial draft, subsequent revisions; SCMV, MSA: case management, review of the manuscript, corrections, and final approval; MSA will act as a case-report guarantor. All authors approved the final version of this manuscript and are responsible for all aspects of this study.
Funding: None; Competing interests: None stated.
References
- Bohan A, Peter JB, Bowman RL, Pearson CM. A computer-assisted analysis of 153 patients with polymyositis and dermatomyositis. Medicine (Baltimore). 1977;56(4):255-286.
- Oka M, Raasakka T. Cardiac involvement in polymyositis. Scand J Rheumatol. 1978;7(4):203-208.
- Odabasi Z, Yapundich R, Oh SJ. Polymyositis presenting with cardiac manifestations: Report of two cases and review of the literature. Clin Neurol Neurosurg. 2010;112(2):160-163.
- Zhang L, Wang G-C, Ma L, Zu N. Cardiac involvement in adult polymyositis or dermatomyositis: a systematic review. Clin Cardiol. 2012;35(11):686-691.
- Jayakumar D, Zhang R, Wasserman A, Ash J. Cardiac manifestations in idiopathic inflammatory myopathies: An overview. Cardiol Rev. 2019;27(3):131-137.
- Allanore Y, Vignaux O, Arnaud L, Puéchal X, Pavy S, Duboc D, et al. Effects of corticosteroids and immunosuppressors on idiopathic inflammatory myopathy related myocarditis evaluated by magnetic resonance imaging. Ann Rheum Dis. 2006;65(2):249-252.
- Yoshimatsu Y, Kotani T, Fujiki Y, Oda K, Kataoka T, Yamairi K, et al. Successful treatment with intravenous high-dose immunoglobulin for cardiomyopathy in dermatomyositis complicated with rapid progressive interstitial pneumonia. Int J Rheum Dis. 2019;22(2):321-324.