Nabaneet Majumder, Abdul Wahid Ayubi, Seema S More, Rohit Kalani1, Sunayana Pawaskar
From the Department of Pathology and Department of Pediatrics1, DY Patil Medical College,
Kolhapur - 416006, India.
Corresponding Author:
Dr. Nabaneet Majumder
Email: drnabaneetmajumder@gmail.com
Abstract
Wolman’s disease is a rare, autosomal recessive, lysosomal storage disease caused by absence or deficiency of lysosomal acid lipase. This leads to accumulation of cholesterol esters and triglycerides in multiple organs of the body. We report an rare case of Wolman’s disease, diagnosed on the basis of acid lipase enzyme estimation and characteristic clinical presentation.
|
6go6ckt5b8|3000F7576AC3|Tab_Articles|Fulltext|0xf1fff49402000000b801000001000500 6go6ckt5b5idvals|184 6go6ckt5b5idcol1|ID 6go6ckt5b5|2000F757Tab_Articles|Fulltext Introduction
Wolman’s disease is an inherited metabolic disorder resulting from the deficiency of lysosomal acid lipase and the accumulation of cholesterol esters and triglycerides in various tissue parts. Lysosomal acid lipase is an essential enzyme that hydrolyzes triglycerides and cholesteryl esters in lysosomes. Mutations in the human LAL gene cause two distinct phenotypes: Wolman disease, a fatal autosomal recessive form and cholesteryl ester storage disease which is a benign adult form [1,2]. Wolman’s disease usually manifests in the first year of life with failure to thrive, relentless vomiting and hepatosplenomegaly. Calcification of the adrenals, foamy macrophages, hepatosplenomegaly is pathognomic for the disease [3]. We here in report one such rare case of Wolman’s disease in a two month old infant.
Case Report
A two month old full term female baby born of non-consanguineous marriage, presented with history of repeated diarrhea, vomiting, progressive abdominal distension and shortness of breath since last three weeks. There was no history suggestive of antenatal infection and birth weight of child was 1.8 kg. There is a previous history of two male infants born of the same couple, who had suffered from similar kind of complaints and expired within first year of life. Examination revealed pale child having firm and nontender hepatosplenomegaly (Liver: 8 cm below right costal margin and spleen: 5 cm in long axis) with no lymphadenopathy or developmental delay.
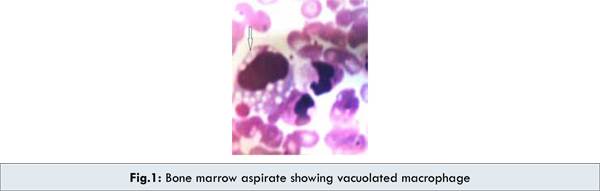
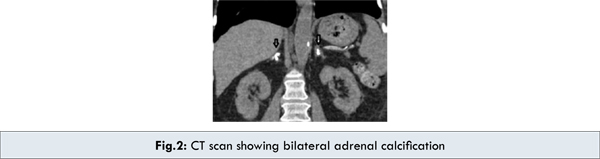
Investigations revealed pancytopenia: hemoglobin 8.2 g/dL, leucocyte count 3,300/cu.mm, platelet count 65 x 109/L. Peripheral blood smear showed a microcytic hypochromic blood picture with few vacuolated lymphocytes. Bone marrow aspirate revealed foamy macrophages [Fig.1]. The liver functions were deranged with elevated liver enzymes: SGOT- 112 U/L (normal range: 5-40 U/L), SGPT- 104 U/L (normal range: 5-42 U/L). Serum Bilirubin 2.1 mg/dL with a direct component of 1.9 mg/dL, (normal range: Total: 0.3-1.0 mg/dL, Direct: 0-0.2 mg/dL). Serum total protein 7gm/dL (normal range: 5.5-8 gm/dL) and serum albumin 3.6 gm/dL (normal range: 3.5-5.0 gm/dL) were within normal limits. Both prothrombin time [40 seconds (normal range: <15 seconds)] and partial thromboplastin time [90 seconds (normal range: <36 seconds)] were prolonged with INR of 3. Serum cholesterol 248 mg/dL (normal range: 140-240 mg/dL) and triglycerides 512 mg/dL (normal range: 25-160 mg/dL) were raised. Chest X-ray and stool examination was normal. Abdominal ultrasound and CT scan showed bilateral adrenal calcifications [Fig.2], with hepatosplenomegaly.
An initial diagnosis of Wolman’s disease was suspected on the basis of family history and features of hepatosplenomegaly, foamy macrophages and bilateral adrenal calcification. However, the diagnosis was confirmed with leukocyte and fibroblast acid lipase enzyme estimation which showed markedly reduced enzyme level of less than 1% of normal. Line of treatment was supportive; eventually the baby expired at the age of four months.
Discussion
Wolman’s disease was first described by Abramov, Schorr and Wolman in 1956 [4,5] . Patrick and Lake in 1969 demonstrated the deficiency of acid lipase for the first time [5]. Wolman’s disease is an infantile-onset disorder with an incidence of <1/300,000 live births and the mean life span for patients with Wolman’s disease is approximately 6 months [1]. The biochemical defect is in the lysosomal acid cholesteryl hydrolase (EC 3.1.1.13), the gene of which is located on chromosome 10q23.2-q23.3.7. Several mutations have been identified, some of which lead to almost complete loss of enzyme activity and to the phenotype of Wolman’s disease, while other mutations cause only partial disruption of the enzyme action, leading to a clinically more benign disease, the so-called cholesterol ester storage disease. In Wolman’s disease, the LDL triglycerides, triglycerides and cholesteryl esters that enter the cells cannot be hydrolyzed, due to the deficiency of lysosomal acid lipase. The consequence of disrupted acid lipase action appears mainly in the adrenal glands, reticulo-endothelial system and intestinal mucosa. A constant feature of the disease in adrenal gland is focal calcification due to necrosis of adrenocortical cells that are overloaded with hydrophobic lipids along with foamy macrophages, hepatosplenomegaly which is pathognomic for the disease. However, the definite diagnosis can only be reached by measuring the acid lipase enzyme activity in leukocytes and cultured skin fibroblasts [3,6].
Our case presented with pancytopenia, hepatosplenomegaly and radiological finding of bilateral adrenal calcification along with foamy macrophages in bone marrow. These features raised the suspicion of Wolman’s disease initially but it had to be differentiated from Niemann-Pick disease, Hypercholesterolemia and Histiocytosis. Other differentials like Adrenal tumors, Histoplasmosis, Tuberculosis and Addison disease were also considered [2,3]. However, the final diagnosis of Wolman’s disease was confirmed in our case with the acid lipase enzyme report which showed markedly reduced enzyme levels ( < 1% of normal).
There is no specific treatment available for Wolman disease [2,5]. Wolman disease has a progressive down hill course eventually leading to death by 3 to 6 months [5]. The main goal of treatment in Wolman’s Disease is to manage the gastrointestinal symptoms of diarrhea and vomiting [2]. A case of Wolman disease successfully treated by bone marrow transplantation has been reported [7]. Prenatal diagnosis by amniocentesis and chorionic villus biopsy [5,8,9] can be done and the affected foetus can be aborted accordingly.
Conclusion
We have highlighted the hallmark clinical presentations of Wolman’s disease. However, a diagnosis of Wolman disease should always be entertained in the evaluation of any child presenting with features of hepatosplenomegaly, foamy histiocytes and adrenal calcification.
Consent:
Parents of the patient have given their informed consent for the case report to be published.
References
- Hong DU, Terri LC, Stephen JG, Gregory PP, Lee AH, Earl W, Kathleen MH, Gregory AG. Wolman disease/cholesteryl ester storage disease efficacy of plant produced human lysosomal acid lipase in mice. J Lipid Res. 2008;49:1646–1657.
- Yousef TM, Shafik MH, Shalan YAF. Wolman Disease in an Egyptian Patient. Kuwait Medical Journal. 2005;37:200-202.
- Panchagnula R, Britto C, Vinod J, Anuradha S, Damodar P. Wolman’s Disease - A Case Report. Indian J. Pathol. Microbiol. 2000;43:91-92.
- Melvin IM, Arthur JM. Wolman’s Disease. Canad Med Ass J. 1968;99.
- SurveTY, Muranjan MN, Barucha BA. Wolman Disease: Diagnosis by Leucocyte Acid Lipase Estimation. Indian J Pediatr. 2005;72:353-354.
- Essa MA, Nounou R, Sakati N, Quesne GL, Joshi S, Archibald A, Ozand PT. Wolman’s Disease: The King Faisal Specialist Hospital And Research Centre Experience. Ann Saudi Med. 1998;18:120-124.
- Krivit W, Peters C, Dusenbery K, Ben YY, Ramsay NKC, Wagner JE, Anderson R. Wolman disease successfully treated by bone marrow transplantation. Bone Marrow Transplantation. 2000;26:567-570.
- Patrick AD, Willcox P, Stephens R, Kenyont VG. Prenatal diagnosis of Wolman’s disease. Journal of Medical Genetics. 1976;13:49-51.
- Coates PM, Cortner JA, Mennuti MT, Wheeler JE. Prenatal diagnosis of Wolman disease. Am J Med Genet. 1978;2:397-407.
|