Introduction
Voltage-gated potassium channel (VGKC) complexes are located on the neuronal membranes of both the central and peripheral nervous systems. They play a crucial role in returning neurons to their resting state after an action potential. There are three primary VGKC-associated subgroups: patients with anti-leucine-rich glioma-inactivated 1 (LGI-1) antibodies, those with anti-contactin-associated protein-like 2 (CASPR2) antibodies, and VGKC-positive patients who lack both antibodies.
CASPR2 antibody encephalitis typically affects elderly males and presents with a more variable spectrum of central nervous system symptoms-such as encephalopathy, hallucinations, seizures, autonomic dysfunction, and insomnia-as well as peripheral nervous system manifestations like neuropathy, fasciculations, and cramps. In contrast, LGI1 antibody encephalitis, which also affects elderly males (median age 60 years), is characterized by memory loss, confusion, and seizures [1]. It is frequently associated with hyponatremia and faciobrachial dystonic seizures, a distinct form of brief tonic or myoclonic-like episodes [2]. Case series have demonstrated the benefits of immunotherapy in both LGI1 and CASPR2 antibody-mediated diseases. Early diagnosis and tumor screening are critical for the effective management of VGKC encephalitis. In this paper, we report four cases of VGKC encephalitis, highlighting their clinical features, atypical MRI findings, treatment approaches, and prognosis.
Case Series
Case 1: CASPR2 Antibody Encephalitis Presenting as Bilateral Temporal Hyperintensity (BTH)
A 60-year-old woman presented with acute behavioural changes, including irritability, forgetfulness, and confusion, followed by altered level of consciousness over 2-3 days. A week prior, she experienced a mild fever, cough, and fatigue, which resolved spontaneously. She had no history of seizures, hallucinations, or focal neurological symptoms. On examination, she was conscious but disoriented, with brisk deep tendon reflexes and withdrawal on plantar stimulation. Primary lab investigations, including CSF analysis and EEG, were normal, except for movement artifacts. MRI revealed bilateral temporo-parietal T2/FLAIR hyperintensities [Fig.1] and T1 hypointensity, with diffusion restriction in the right insular cortex.
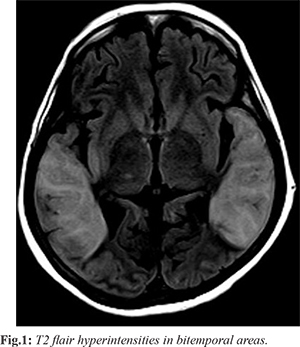
Herpes encephalitis was suspected, and she received a 14-day course of acyclovir, but there was no improvement. Subsequent autoimmune testing revealed positive anti-CASPR2 antibodies, confirming the diagnosis of CASPR2 antibody encephalitis. Due to financial constraints, paraneoplastic screening and more aggressive therapies such as IVIG and plasmapheresis were deferred. Despite treatment with high-dose steroids and cyclophosphamide, the patient showed minimal improvement in sensorium. After discharge, follow-up was recommended, but contact was lost in July 2021.
Case 2: LGI1 Antibody Encephalitis with Gyriform Hyperintensity
A 56-year-old man presented with a one-month history of acute-onset giddiness, falls, involuntary movements, fever, and altered sensorium. His symptoms began with brief, frequent episodes of giddiness, which escalated into visual hallucinations, behavioral changes, and memory loss. By the third week, the patient developed high-grade fever and frequent myoclonic jerks, which worsened despite symptomatic treatment. He had a history of an upper respiratory tract infection two months prior.
On admission, his vitals were stable, but he was disoriented with brisk deep tendon reflexes. CSF analysis revealed lymphocytic pleocytosis, and MRI showed bilateral temporal lobe and hippocampal hyperintensities, suggestive of HSV encephalitis [Fig.2]. Despite a week of acyclovir therapy, there was no improvement. Further testing confirmed LGI1 antibody encephalitis. The patient was treated with pulse steroids and IVIG, which led to a reduction in myoclonic jerks and improved his sensorium. Follow-up imaging and investigations revealed no malignancy. He was discharged on oral steroids, azathioprine, and antiepileptics, and showed steady improvement over the following months, with a modified Rankin Score (mRS) of 5 at three and six months.
Case 3: LGI1 Encephalitis with Hyponatremia and Medial Temporal Lobe Hyperintensity
A 68-year-old man with no significant past medical history presented with progressive memory impairment over 1.5 months, along with a single episode of tonic-clonic seizures. His symptoms were preceded by two days of fever and myalgia. He had no history of myoclonus, visual disturbances, or focal neurological deficits. On examination, he was inattentive, with poor cognitive scores on both the MMSE (7/30) and MOCA (8/30). His neurological examination was otherwise unremarkable.
Initial lab results revealed hyponatremia (serum sodium of 121 mEq/L), but his other blood work was within normal limits. CSF analysis showed mild lymphocytic pleocytosis, and MRI revealed bilateral medial temporal lobe T2/FLAIR hyperintensities [Fig.3,4]. Autoimmune panel testing confirmed LGI1 antibody positivity, and he was treated with pulse steroids followed by oral steroids. His caregivers noted marked improvement in his orientation and behavior, with his MMSE improving to 17/30 by discharge. At six-month follow-up, his MRS was 2, reflecting a significant recovery.
Case 4: LGI1 Encephalitis with Normal MRI and Extreme Delta Brush
A 55-year-old waiter presented with six months of recurrent seizures affecting his upper and lower limbs, initially controlled with antiepileptic medication. However, over the last two weeks, his seizures recurred with increasing frequency, culminating in 20 episodes without recovery of consciousness just before admission. There was no history of behavioral changes, focal neurological deficits, or systemic symptoms such as fever or weight loss.
Upon admission, the patient was in status epilepticus, requiring intubation and sedation. His initial labs were unremarkable, and MRI showed no parenchymal abnormalities. EEG revealed an abnormal pattern of extreme delta brush activity [Fig.5]. Testing for autoimmune encephalitis confirmed the presence of LGI1 antibodies. He was treated with pulse steroids, antiseizure medications, and empirical antibiotics. Despite these efforts, the patient developed refractory shock and sepsis, and he died on the fifth day of admission.
These cases highlight the diverse presentations and imaging findings of VGKC-associated limbic encephalitis, with emphasis on the challenges of timely diagnosis and the importance of early immunotherapy.
Discussion
The clinical and imaging features of voltage-gated potassium channel (VGKC)-associated limbic encephalitis (LE) are constantly evolving. Antibodies targeting VGKC complexes can result in a variety of neurological manifestations, with LE being the most frequent presentation, followed by neuromyotonia, Morvan’s syndrome, epilepsy, and other uncategorized central nervous system (CNS) features [
3].
In the first case, we reported the presence of CASPR2 antibodies in a female patient presenting with acute delirium and bilateral temporal lobe hyperintensities. To our knowledge, this is one of the few cases describing CASPR2 antibody-associated LE with such extensive bilateral temporal lobe involvement. Bilateral temporal lobe hyperintensities (BTH) have a limited set of differential diagnoses, including herpes encephalitis (the most common cause), mesial temporal sclerosis, gliomatosis cerebri, and certain neurodegenerative diseases [4]. While herpes encephalitis is the most frequent cause of BTH, CASPR2-associated autoimmune encephalitis presenting as BTH is rare and not widely reported in the literature [5]. Interestingly, CASPR2 encephalitis is more common in males, with Morvan's syndrome and LE being the most prevalent presentations [6]. Although CASPR2 antibody encephalitis can mimic other forms of autoimmune encephalitis, a thorough workup ruled out alternative causes, and the clinical and radiological findings strongly supported an autoimmune etiology. Antibody titers were unavailable, but given the clinical context, a false positive CASPR2 result was deemed unlikely [7]. The second case emphasizes the classical presentation of LGI1 antibody encephalitis, characterized by prodromal symptoms, clinical features, and MRI findings. However, an unusual aspect of this case was the presence of cortical laminar necrosis, a less frequent finding in LGI1 encephalitis. Typically, VGKC encephalitis is associated with MRI findings such as T2/FLAIR hyperintensities, diffusion restriction, and post-contrast enhancement in the temporal lobes, often progressing to mesial temporal lobe sclerosis [8]. Cortical laminar necrosis is pathologically defined as the selective necrosis of specific cortical layers, affecting neurons, glial cells, and cerebral blood vessels [9]. Although it is most often caused by metabolic disorders, hypoxia, and conditions like hypoglycemia or hepatic/renal dysfunction, cortical laminar necrosis can also occur in encephalitis [10,11].
Conclusion
VGKC encephalitis presents with diverse clinical and imaging findings, making early recognition critical for improving patient outcomes. Understanding the broad spectrum of neurological symptoms and radiological features, such as bilateral temporal lobe hyperintensities and rare findings like cortical laminar necrosis, is key to preventing misdiagnosis and ensuring prompt, appropriate treatment to prevent long-term disability.
Contributors: AS: manuscript writing, literature search; PVK: manuscript editing, patient management. PVK will act as a study guarantor. Both authors approved the final version of this manuscript and are responsible for all aspects of this study.
Funding: None; Competing interests: None stated.
References
- Tüzün E, Dalmau J. Limbic encephalitis and variants: Classification, diagnosis and treatment. Neurologist 2007;13:261-271.
- Lai M, Hughes EG, Peng X, Zhou L, Gleichman AJ, Shu H, et al. AMPA receptor antibodies in limbic encephalitis alter synaptic receptor location. Ann Neurol. 2009;65:424-434.
- Vincent A, Bien CG, Irani SR, Waters P. Autoantibodies associated with diseases of the CNS: New developments and future challenges. Lancet Neurol. 2011;10:759-772.
- Sureka J, Jakkani RK. Clinico-radiological spectrum of bilateral temporal lobe hyperintensity: a retrospective review. Br J Radiol. 2012;85(1017):e782-e792.
- van Sonderen A, Thijs RD, Coenders EC, Jiskoot LC, Sanchez E, de Bruijn MA, et al. Anti-LGI1 encephalitis: clinical syndrome and long-term follow-up. Neurology. 2016;87:1449-1456.
- Boyko M, Au KLK, Casault C, de Robles P, Pfeffer G. Systematic review of the clinical spectrum of CASPR2 antibody syndrome. J Neurol. 2020;267(4):1137-1146.
- Bien CG. Overinterpretation and overtreatment of low-titer antibodies against Contactin-associated protein-2. Front Immunol. 2018;9:703.
- Kotsenas AL, Watson RE, Pittock SJ, Britton JW, Hoye SL, Quek AML, et al. MRI findings in autoimmune voltage-gated potassium channel complex encephalitis with seizures: one potential etiology for mesial temporal sclerosis. Am J Neuroradiol. 2014;35:84-89.
- Garg RK, Rizvi I, Ingole R, Jain A, Malhotra HS, Kumar N, et al. Cortical laminar necrosis in dengue encephalitis-a case report. BMC Neurol. 2017;17(1):79.
- Niwa T, Aida N, Shishikura A, Fujita K, Inoue T. Susceptibility-weighted imaging findings of cortical laminar necrosis in pediatric patients. Am J Neuroradiol. 2008;29(9):1795-1798.
- Rajasekharan C, Jithesh B, Renjith SW. Cortical laminar necrosis due to hypoglycaemic encephalopathy - images in medicine. BMJ Case Rep. 2013:bcr2012007726.